
[Edited Post]
As this link https://labs.imaginea.com/novel-corona-virus-sequence-alignment/ stopped working, so I have used http://web.archive.org/ to recreate the blog and repost it here.
Happy reading.
Pairwise sequence alignment is a useful tool in many fields of biology. For example, the similarity between sequences can used be in evolutionary analysis to find out what organisms share a common ancestor. Alternatively, the similarity between amino acid sequences can be used to predict the structure and functions of protein domains. Another use of pairwise sequence analysis is in genome sequencing assembly, where matches are used to find overlaps in the shorter pieces of DNA sequenced. (Source:Genome Assembly Page)
Now the question we will try to answer in the post:
How similar are two sequences? Let us know some terms before we jump into answering the above question with coding and visualizing the sequence.
What is Sequence Alignment ?
Sequence alignment is a technique for arranging sequences of DNA, RNA, or protein to identify sectors/regions of similarity. It is a part of Genome Assembly which you can read about at this post.
The similarity of the two given sequence may result of structural or evolutionary or functionality similarity.
How does a Genome Look?
I will be using nucleotide sequencing of Coronavirus and try to compare it with a common existing virus like Human Rhinovirus A1.
Below is the example of Coronavirus Nucleotide:
cacgcagtat aattaataac taattactgt cgttgacagg acacgagtaa ctcgtctatc
Here, cytosine (c) is followed by adenine (a) which is followed by a cytosine (c), which in turn is followed by a guanine (g), and so on. (T stands for thymine(t))
Here, I will be using Pairwise Similarity Alignment and see the scores.
How does Pairwise Similarity Alignment work ?
Let us see the below image and see different possible components of a sequence.

There can be multiple ways to score the similarity from the genome sequence, like,
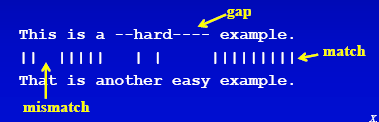

We can chalk down 3 basic aspects for scoring.
Match Value Scoring -Assign a value when there is an exact math Mismatch Value Scoring -Generally a negative value is assigned if there is a mismatch. Gap Penalty - Gaps can be either Open or Extended gaps which is also assigned negative values.
Types of Pairwise Similarity Alignment
We will take two sequence $X$ and $Y$,
\[X = x_0 ... x_i ... x_n \\ Y = y_0 ... y_i ... y_n\]I will be using BioPython for this example.
If someone wants to use a practice sequence then it can be used as well.
# practice sequences to be aligned
X = "ACGGGT"
Y = "ACG"
Global Alignment
It helps find the best alignment over the entire lengths of the 2 sequences $X$ and $Y$.
What is the maximum similarity between sequence $X$ and $Y$?
Binary Global Alignment
If both the sequence is exactly same then and Match Value Scoring = 1, Mismatch Value Scoring = 0 and Gap Penalty is not considered then the absolute score will be.
Code:
from Bio import pairwise2
from Bio.pairwise2 import format_alignment
# Define two sequences to be aligned (X==Y)
X = "attaaaggtt tataccttcc caggtaacaa accaaccaac tttcgatctc ttgtagatct"
Y = "attaaaggtt tataccttcc caggtaacaa accaaccaac tttcgatctc ttgtagatct"
# No parameters. Identical characters have score of 1, else 0.
# No gap penalties.
al = pairwise2.align.globalxx(X, Y)
# Use format_alignment method to format the alignments in the list
for a in al:
print(format_alignment(*a))
Output:
attaaaggtt tataccttcc caggtaacaa accaaccaac tttcgatctc ttgtagatct
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
attaaaggtt tataccttcc caggtaacaa accaaccaac tttcgatctc ttgtagatct
Score=65
If it is exactly same then the score is 65.
Now, let us take two different sequence one from Coronavirus and another from Human Rhinovirus A1 of the first sequence.
Code:
from Bio import pairwise2
from Bio.pairwise2 import format_alignment
# Define two sequences to be aligned (X==Y)
X = "attaaaggtt tataccttcc caggtaacaa accaaccaac tttcgatctc ttgtagatct"
Y = "attaaaggtt tataccttcc caggtaacaa accaaccaac tttcgatctc ttgtagatct"
# No parameters. Identical characters have score of 1, else 0.
# No gap penalties.
al = pairwise2.align.globalxx(X, Y)
# Use format_alignment method to format the alignments in the list
for a in al:
print(format_alignment(*a))
Output:
attaaa---gg--t-t ---tatacc--ttcc caggtaaca--a -accaacc-aac t---t-tc-gat--ctc ttgtagatc-t
||||| || | | | | |||| | || || ||| ||| || || | | | | ||| ||| | |
-ttaaaactgg gtgt-gggt-t---g ttcc-c-----ac-cca cacc-acccaa- tgggtgt-tg-t actc--tgt---t-at
Score=42
attaa-a--gg--t-t ---tatacc--ttcc caggtaaca--a -accaacc-aac t---t-tc-gat--ctc ttgtagatc-t
|||| | || | | | | |||| | || || ||| ||| || || | | | | ||| ||| | |
-ttaaaactgg gtgt-gggt-t---g ttcc-c-----ac-cca cacc-acccaa- tgggtgt-tg-t actc--tgt---t-at
Score=42
atta-aa--gg--t-t ---tatacc--ttcc caggtaaca--a -accaacc-aac t---t-tc-gat--ctc ttgtagatc-t
||| || || | | | | |||| | || || ||| ||| || || | | | | ||| ||| | |
-ttaaaactgg gtgt-gggt-t---g ttcc-c-----ac-cca cacc-acccaa- tgggtgt-tg-t actc--tgt---t-at
Score=42
att-aaa--gg--t-t ---tatacc--ttcc caggtaaca--a -accaacc-aac t---t-tc-gat--ctc ttgtagatc-t
|| ||| || | | | | |||| | || || ||| ||| || || | | | | ||| ||| | |
-ttaaaactgg gtgt-gggt-t---g ttcc-c-----ac-cca cacc-acccaa- tgggtgt-tg-t actc--tgt---t-at
Score=42
...
attaaa----g-gt-t-- tatacc--ttcc caggtaaca--a -accaacc-aac t---t-tc-gat--ctc ttgtagatc-t
||||| | || | .| | |||| | || || ||| ||| || || | | | | ||| ||| | |
-ttaaaactgg gtgtgggt-t---g ttcc-c-----ac-cca cacc-acccaa- tgggtgt-tg-t actc--tgt---t-at
Score=42
attaa-a---g-gt-t-- tatacc--ttcc caggtaaca--a -accaacc-aac t---t-tc-gat--ctc ttgtagatc-t
|||| | | || | .| | |||| | || || ||| ||| || || | | | | ||| ||| | |
-ttaaaactgg gtgtgggt-t---g ttcc-c-----ac-cca cacc-acccaa- tgggtgt-tg-t actc--tgt---t-at
Score=42
atta-aa---g-gt-t-- tatacc--ttcc caggtaaca--a -accaacc-aac t---t-tc-gat--ctc ttgtagatc-t
||| || | || | .| | |||| | || || ||| ||| || || | | | | ||| ||| | |
-ttaaaactgg gtgtgggt-t---g ttcc-c-----ac-cca cacc-acccaa- tgggtgt-tg-t actc--tgt---t-at
Score=42
att-aaa---g-gt-t-- tatacc--ttcc caggtaaca--a -accaacc-aac t---t-tc-gat--ctc ttgtagatc-t
|| ||| | || | .| | |||| | || || ||| ||| || || | | | | ||| ||| | |
-ttaaaactgg gtgtgggt-t---g ttcc-c-----ac-cca cacc-acccaa- tgggtgt-tg-t actc--tgt---t-at
Score=42
Here we can see a average score of 42 out of 65.
Weighted Global Alignment
Here, each and every possible type of scoring is given a weight.
If both the sequence is exactly same then and Match Value Scoring = 2, Mismatch Value Scoring = -1 and Gap Penalty (Open=-0.5, Extend=-0.1) then the weighted score will be.
Code:
from Bio import pairwise2
from Bio.pairwise2 import format_alignment
# Define two sequences to be aligned (X==Y)
X = "attaaaggtt tataccttcc caggtaacaa accaaccaac tttcgatctc ttgtagatct"
Y = "attaaaggtt tataccttcc caggtaacaa accaaccaac tttcgatctc ttgtagatct"
# match is the score to given to identical characters.
# mismatch is the score given to non-identical ones.
# open and extend are the gap penalties when a gap is
# opened and extended. They should be negative.
al = pairwise2.align.globalms(X, Y, 2, -1, -0.5, -0.1)
# Use format_alignment method to format the alignments in the list
for a in al:
print(format_alignment(*a))
Output:
attaaaggtt tataccttcc caggtaacaa accaaccaac tttcgatctc ttgtagatct
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
attaaaggtt tataccttcc caggtaacaa accaaccaac tttcgatctc ttgtagatct
Score=130
If it is a exact match then the score is 130.
Now lets plugin the data from Rhinovirus and see the results for weighted global alignment.
Code:
from Bio import pairwise2
from Bio.pairwise2 import format_alignment
# Define two sequences to be aligned (X==Y)
X = "attaaaggtt tataccttcc caggtaacaa accaaccaac tttcgatctc ttgtagatct"
Y = "ttaaaactgg gtgtgggttg ttcccaccca caccacccaa tgggtgttgt actctgttat"
# match is the score to given to identical characters.
# mismatch is the score given to non-identical ones.
# open and extend are the gap penalties when a gap is
# opened and extended. They should be negative.
al = pairwise2.align.globalms(X, Y, 2, -1, -0.5, -0.1)
# Use format_alignment method to format the alignments in the list
for a in al:
print(format_alignment(*a))
Output:
attaaa-----------ggtt- tataccttc-c caggtaacaa -accaacc-aac t-----ttcg--atctc ttgt-agatct
||||| |||| || | || | | | | || ||| ||| || || || | | ||| ||| | |
-ttaaaactgg gtgtgggttg t-t-cc--cac-c------c-a cacc-acccaa- tgggtgtt-gt a-ctc--tgtta----t
Score=71
attaaa-----------ggtt- tataccttc-c caggtaacaa -accaacc-aac t-----ttcg--atctc ttgt-agatct
||||| |||| || | || | | | || | ||| ||| || || || | | ||| ||| | |
-ttaaaactgg gtgtgggttg t-t-cc--cac-c------ca- cacc-acccaa- tgggtgtt-gt a-ctc--tgtta----t
Score=71
attaaa-----------ggtt- tataccttc-c caggtaacaa -accaacc-aac t-----ttcg--atctc ttgt-agatct
||||| |||| || | || | | | | || |||| || || || || | | ||| ||| | |
-ttaaaactgg gtgtgggttg t-t-cc--cac-c------c-a cacca-cccaa- tgggtgtt-gt a-ctc--tgtta----t
Score=71
attaaa-----------ggtt- tataccttc-c caggtaacaa -accaacc-aac t-----ttcg--atctc ttgt-agatct
||||| |||| || | || | | | || | |||| || || || || | | ||| ||| | |
-ttaaaactgg gtgtgggttg t-t-cc--cac-c------ca- cacca-cccaa- tgggtgtt-gt a-ctc--tgtta----t
Score=71
...
attaaa-----------ggtt- tataccttc-c caggtaacaa -accaa-ccaac t-----ttcgat--ctc ttg-tagatct
||||| |||| || | || | | | | || |||| |||| || || | | ||| || || |
-ttaaaactgg gtgtgggttg t-t-cc--cac-c------c-a cacca-cccaa- tgggtgtt-g-t actc--tgtta----t
Score=71
attaaa-----------ggtt- tataccttc-c caggtaacaa -accaa-ccaac t-----ttcgat--ctc ttg-tagatct
||||| |||| || | || | | | || | |||| |||| || || | | ||| || || |
-ttaaaactgg gtgtgggttg t-t-cc--cac-c------ca- cacca-cccaa- tgggtgtt-g-t actc--tgtta----t
Score=71
attaaa-----------ggtt- tataccttc-c caggtaacaa -accaaccaac t-----ttcgat--ctc ttg-tagatct
||||| |||| || | || | | | | || ||||.|||| || || | | ||| || || |
-ttaaaactgg gtgtgggttg t-t-cc--cac-c------c-a caccacccaa- tgggtgtt-g-t actc--tgtta----t
Score=71
attaaa-----------ggtt- tataccttc-c caggtaacaa -accaaccaac t-----ttcgat--ctc ttg-tagatct
||||| |||| || | || | | | || | ||||.|||| || || | | ||| || || |
-ttaaaactgg gtgtgggttg t-t-cc--cac-c------ca- caccacccaa- tgggtgtt-g-t actc--tgtta----t
Score=71
We can see an average score of 71 out of 130.
There is one more way we can do an alignment i.e. a local alignment.
Local Alignment
This helps to find the most similar subsequences among the given two sequences.
What is the maximum similarity between a subsequence of $X$ and a subsequence of $Y$?
Binary Local Alignment
If both the sequence is exactly same then and Match Value Scoring = 1, Mismatch Value Scoring = 0 and Gap Penalty is not considered then the absolute score will be.
Code:
from Bio import pairwise2
from Bio.pairwise2 import format_alignment
# Define two sequences to be aligned (X==Y)
X = "attaaaggtt tataccttcc caggtaacaa accaaccaac tttcgatctc ttgtagatct"
Y = "attaaaggtt tataccttcc caggtaacaa accaaccaac tttcgatctc ttgtagatct"
# No parameters. Identical characters have score of 1, else 0.
# No gap penalties.
al = pairwise2.align.localxx(X, Y)
# Use format_alignment method to format the alignments in the list
for a in al:
print(format_alignment(*a))
Output:
attaaaggtt tataccttcc caggtaacaa accaaccaac tttcgatctc ttgtagatct
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
attaaaggtt tataccttcc caggtaacaa accaaccaac tttcgatctc ttgtagatct
Score=65
So, if the sequence is exactly same then the score is exactly same as global i.e. 65. But we are not expecting a different score when sequence $Y$ is changed as combinations and the change position will be same.
Code:
from Bio import pairwise2
from Bio.pairwise2 import format_alignment
# Define two sequences to be aligned (X==Y)
X = "attaaaggtt tataccttcc caggtaacaa accaaccaac tttcgatctc ttgtagatct"
Y = "ttaaaactgg gtgtgggttg ttcccaccca caccacccaa tgggtgttgt actctgttat"
# No parameters. Identical characters have score of 1, else 0.
# No gap penalties.
al = pairwise2.align.localxx(X, Y)
# Use format_alignment method to format the alignments in the list
for a in al:
print(format_alignment(*a))
Output:
2 ttaaa---gg--t-t ---tatacc--ttcc caggtaaca--a -accaacc-aac t---t-tc-gat--ctc ttgtagatc-t
||||| || | | | | |||| | || || ||| ||| || || | | | | ||| ||| | |
1 ttaaaactgg gtgt-gggt-t---g ttcc-c-----ac-cca cacc-acccaa- tgggtgt-tg-t actc--tgt---t-at
Score=42
2 ttaa-a--gg--t-t ---tatacc--ttcc caggtaaca--a -accaacc-aac t---t-tc-gat--ctc ttgtagatc-t
|||| | || | | | | |||| | || || ||| ||| || || | | | | ||| ||| | |
1 ttaaaactgg gtgt-gggt-t---g ttcc-c-----ac-cca cacc-acccaa- tgggtgt-tg-t actc--tgt---t-at
Score=42
2 tta-aa--gg--t-t ---tatacc--ttcc caggtaaca--a -accaacc-aac t---t-tc-gat--ctc ttgtagatc-t
||| || || | | | | |||| | || || ||| ||| || || | | | | ||| ||| | |
1 ttaaaactgg gtgt-gggt-t---g ttcc-c-----ac-cca cacc-acccaa- tgggtgt-tg-t actc--tgt---t-at
Score=42
2 tt-aaa--gg--t-t ---tatacc--ttcc caggtaaca--a -accaacc-aac t---t-tc-gat--ctc ttgtagatc-t
|| ||| || | | | | |||| | || || ||| ||| || || | | | | ||| ||| | |
1 ttaaaactgg gtgt-gggt-t---g ttcc-c-----ac-cca cacc-acccaa- tgggtgt-tg-t actc--tgt---t-at
Score=42
...
2 ttaaa----g-gt-t-- tatacc--ttcc caggtaaca--a -accaacc-aac t---t-tc-gat--ctc ttgtagatc-t
||||| | || | .| | |||| | || || ||| ||| || || | | | | ||| ||| | |
1 ttaaaactgg gtgtgggt-t---g ttcc-c-----ac-cca cacc-acccaa- tgggtgt-tg-t actc--tgt---t-at
Score=42
2 ttaa-a---g-gt-t-- tatacc--ttcc caggtaaca--a -accaacc-aac t---t-tc-gat--ctc ttgtagatc-t
|||| | | || | .| | |||| | || || ||| ||| || || | | | | ||| ||| | |
1 ttaaaactgg gtgtgggt-t---g ttcc-c-----ac-cca cacc-acccaa- tgggtgt-tg-t actc--tgt---t-at
Score=42
2 tta-aa---g-gt-t-- tatacc--ttcc caggtaaca--a -accaacc-aac t---t-tc-gat--ctc ttgtagatc-t
||| || | || | .| | |||| | || || ||| ||| || || | | | | ||| ||| | |
1 ttaaaactgg gtgtgggt-t---g ttcc-c-----ac-cca cacc-acccaa- tgggtgt-tg-t actc--tgt---t-at
Score=42
2 tt-aaa---g-gt-t-- tatacc--ttcc caggtaaca--a -accaacc-aac t---t-tc-gat--ctc ttgtagatc-t
|| ||| | || | .| | |||| | || || ||| ||| || || | | | | ||| ||| | |
1 ttaaaactgg gtgtgggt-t---g ttcc-c-----ac-cca cacc-acccaa- tgggtgt-tg-t actc--tgt---t-at
Score=42
It produces the same score like Binary Global Alignment. But this is a subsequence matching. It is advised to change the sequence a bit and observe the change it makes.
Weighted Local Alignment
If both the sequence is exactly same then and Match Value Scoring = 2, Mismatch Value Scoring = -1 and Gap Penalty (Open=-0.5, Extend=-0.1) then the weighted score will be.
Code:
from Bio import pairwise2
from Bio.pairwise2 import format_alignment
# Define two sequences to be aligned (X==Y)
X = "attaaaggtt tataccttcc caggtaacaa accaaccaac tttcgatctc ttgtagatct"
Y = "attaaaggtt tataccttcc caggtaacaa accaaccaac tttcgatctc ttgtagatct"
# match is the score to given to identical characters.
# mismatch is the score given to non-identical ones.
# open and extend are the gap penalties when a gap is
# opened and extended. They should be negative.
al = pairwise2.align.localms(X, Y, 2, -1, -0.5, -0.1)
# Use format_alignment method to format the alignments in the list
for a in al:
print(format_alignment(*a))
Output:
attaaaggtt tataccttcc caggtaacaa accaaccaac tttcgatctc ttgtagatct
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
attaaaggtt tataccttcc caggtaacaa accaaccaac tttcgatctc ttgtagatct
Score=130
It has the same value of 130 like globalms.
Code:
from Bio import pairwise2
from Bio.pairwise2 import format_alignment
# Define two sequences to be aligned (X==Y)
X = "attaaaggtt tataccttcc caggtaacaa accaaccaac tttcgatctc ttgtagatct"
Y = "ttaaaactgg gtgtgggttg ttcccaccca caccacccaa tgggtgttgt actctgttat"
# match is the score to given to identical characters.
# mismatch is the score given to non-identical ones.
# open and extend are the gap penalties when a gap is
# opened and extended. They should be negative.
al = pairwise2.align.localms(X, Y, 2, -1, -0.5, -0.1)
# Use format_alignment method to format the alignments in the list
for a in al:
print(format_alignment(*a))
Output:
2 ttaaa-----------ggtt- tataccttc-c caggtaacaa -accaacc-aac t-----ttcg--atctc ttgtag-at
||||| |||| || | || | | | | || ||| ||| || || || | | ||| ||| ||
1 ttaaaactgg gtgtgggttg t-t-cc--cac-c------c-a cacc-acccaa- tgggtgtt-gt a-ctc--tgt--tat
Score=71.7
2 ttaaa-----------ggtt- tataccttc-c caggtaacaa -accaacc-aac t-----ttcg--atctc ttgtag-at
||||| |||| || | || | | | || | ||| ||| || || || | | ||| ||| ||
1 ttaaaactgg gtgtgggttg t-t-cc--cac-c------ca- cacc-acccaa- tgggtgtt-gt a-ctc--tgt--tat
Score=71.7
2 ttaaa-----------ggtt- tataccttc-c caggtaacaa -accaacc-aac t-----ttcg--atctc ttgtag-at
||||| |||| || | || | | | | || |||| || || || || | | ||| ||| ||
1 ttaaaactgg gtgtgggttg t-t-cc--cac-c------c-a cacca-cccaa- tgggtgtt-gt a-ctc--tgt--tat
Score=71.7
2 ttaaa-----------ggtt- tataccttc-c caggtaacaa -accaacc-aac t-----ttcg--atctc ttgtag-at
||||| |||| || | || | | | || | |||| || || || || | | ||| ||| ||
1 ttaaaactgg gtgtgggttg t-t-cc--cac-c------ca- cacca-cccaa- tgggtgtt-gt a-ctc--tgt--tat
Score=71.7
...
2 ttaaa-----------ggtt- tataccttc-c caggtaacaa -accaa-ccaac t-----ttcgat--ctc ttg-tagat
||||| |||| || | || | | | | || |||| |||| || || | | ||| || || |
1 ttaaaactgg gtgtgggttg t-t-cc--cac-c------c-a cacca-cccaa- tgggtgtt-g-t actc--tgtta--t
Score=71.7
2 ttaaa-----------ggtt- tataccttc-c caggtaacaa -accaa-ccaac t-----ttcgat--ctc ttg-tagat
||||| |||| || | || | | | || | |||| |||| || || | | ||| || || |
1 ttaaaactgg gtgtgggttg t-t-cc--cac-c------ca- cacca-cccaa- tgggtgtt-g-t actc--tgtta--t
Score=71.7
2 ttaaa-----------ggtt- tataccttc-c caggtaacaa -accaaccaac t-----ttcgat--ctc ttg-tagat
||||| |||| || | || | | | | || ||||.|||| || || | | ||| || || |
1 ttaaaactgg gtgtgggttg t-t-cc--cac-c------c-a caccacccaa- tgggtgtt-g-t actc--tgtta--t
Score=71.7
2 ttaaa-----------ggtt- tataccttc-c caggtaacaa -accaaccaac t-----ttcgat--ctc ttg-tagat
||||| |||| || | || | | | || | ||||.|||| || || | | ||| || || |
1 ttaaaactgg gtgtgggttg t-t-cc--cac-c------ca- caccacccaa- tgggtgtt-g-t actc--tgtta--t
Score=71.7
Here, the score is different from Weighted Global Alignment. The score is 71.5 out of 130.
We can test with other virus RNA’s and find the similarity score. That will help us to give more insight about Coronavirus. There are advanced algorithms which dynamic programming to obtain Global and Local Alignments.
Further Readings
- Smith-Waterman Algorithm
- Convex/Affine Gap Penalty
- Needleman-Wunch Algorithm
References
- http://biopython.org/DIST/docs/tutorial/Tutorial.html
- http://www.genomenewsnetwork.org/resources/whats_a_genome/Chp2_1.shtml
- http://compbio.pbworks.com/w/page/16252912/Pairwise%20Sequence%20Alignment
- https://www.ncbi.nlm.nih.gov/nuccore/MN908947?fbclid=IwAR1tyqtVCorwopIhxSwEkW3rVbWhAIcEbvT75K0p5WEQImflzolX6Yw3f8o
- https://www.ncbi.nlm.nih.gov/nuccore/NC_038311.1
- https://molbiol-tools.ca/Genomics.htm
- https://en.wikipedia.org/wiki/Nucleic_acid_sequence
- http://compbio.pbworks.com/w/page/16252894/Genome%20Sequencing%20and%20Assembly
SAME POST AT Imaginea Labs
[Edited Post]
As this link https://labs.imaginea.com/novel-corona-virus-sequence-alignment/ stopped working, so I have used http://web.archive.org/ to recreate the blog and repost it here. Happy reading.